https://doi.org/10.1038/s41564-023-01426-7Nat Microbiol 8, 1392–1396 (2023).
Human microbiome research has undergone rapid growth over the past two decades and thousands of research papers on this topic are now published every year. Huge sums of money have been spent investigating the human microbiome as a cause of, or potential therapeutic solution to, a wide range of diseases, including inflammatory bowel disease and cardiometabolic conditions. Although truly exciting, the increasing focus on microbiome research has unfortunately also brought with it hype and entrenched certain misconceptions. As a result, many unsupported, or undersupported, statements have become ‘fact’ by virtue of constant repetition. Some are more widespread than others and some are relatively trivial, but, cumulatively, they highlight that misinformation is pervasive in the human microbiome literature. Given the potential importance of human microbiomes for health, it is crucial that claims are based on evidence. In this Perspective, we shine a light on persistent or emerging microbiome myths and misconceptions, outlining factual inaccuracies. We begin with relatively minor, but illustrative, points and build towards issues with greater potential impacts. We have purposefully tried to avoid unnecessary finger pointing at original sources of erroneous information, and instead hope that our insights and critical assessment are helpful to the field.
“Microbiome research is a new field”
The pace of human microbiome research has greatly accelerated over the past 15 years, but the field is not in its infancy. To state so does a disservice to the excellent research that preceded the advent of high-throughput DNA-sequencing approaches. Indeed, there has been a rich history of research into human-associated microorganisms since at least the late nineteenth century. Escherichia coli was first isolated in 18851, bifidobacteria were described in 18992 and Metchnikoff speculated on the importance of beneficial gut microorganisms in the early 1900s3. Similarly, concepts such as the gut–brain axis have been researched for centuries4 and health impacts of key microbiome-associated metabolites, such as short-chain fatty acids, were first reported more than 40 years ago5.
“Joshua Lederberg coined the term ‘microbiome’”
Although Nobel laureate Joshua Lederberg had many notable achievements in his career, he did not invent the word ‘microbiome’. This oft-repeated claim has been thoroughly refuted elsewhere, with evidence provided that the word was used in its modern context more than a decade before Joshua Lederberg first used it in 20016. Although relatively trivial, this myth serves as an example of how easy it is for falsehoods to become embedded in the human microbiome literature.
“There are 1012 bacterial cells per gram of human faeces”
This figure is commonly mentioned in the microbiome literature, but its source has been difficult to ascertain. It may, however, have originated from dry-weight rather than wet-weight faecal cell counts. Regardless, it is incorrect. The real figure, as ascertained using various methods such as direct cell counts, fluorescence in situ hybridization, flow cytometry and quantitative polymerase chain reaction (qPCR), is typically between 1010 and 1011microbial cells per wet-weight gram of faeces7,8,9.
“The human microbiota weighs 1 to 2 kg”
Although this is mentioned many times in the literature, it is often given without citation and we were unable to find an original source for this claim. Nonetheless, it is unlikely to be true in most cases. The majority of the human microbiota resides in the colon, and these microorganisms typically account for less than half of the weight of faecal solids10. The average human stool weighs less than 200 g (wet weight)11, with total colonic contents ranging from 83 to 421 g in a small study of sudden-death victims12. Therefore, aside perhaps from rare cases of severely constipated individuals with extremely compacted faecal matter in their colons, the total weight of the human microbiota is much more likely to be less than 500 g, and perhaps even considerably lower than this in some cases.
“The microbiota outnumbers human cells by 10:1”
This myth is perhaps one of the most pervasive in the human microbiome literature and is one that we have also repeated uncritically in the past (we, sadly, are not immune to mythology). Excellent work10 has, however, shown that this myth seems to have originated from a ‘back of the envelope’ calculation in the 1970s. More detailed analyses indicate that the true figure, albeit still impressive, is probably closer to a ratio of 1:1. It should be noted that the ratio is likely to vary from person to person and is dependent on factors such as the host’s body size and the amount of faecal material they are carrying in their colon13. Current estimates are also largely based on observations from adult individuals living in urbanized high-income country settings. More comprehensive estimates will require study of individuals from lower income or rural settings, and also from across the life course.
“The microbiota is inherited from the mother at birth”
Although variants of this statement are more often found in popular science articles than the scientific literature, it is an example of how nuance is extremely important when describing the human microbiome. Although some microorganisms are directly transferred from mother to baby during birth14,15, proportionally few microbiota species are truly ‘heritable’ and persist through from birth to adulthood in the offspring15,16. Indeed, most of the expansion in gut microbiota diversity occurs after birth, during the first few years of life, and increases most dramatically after weaning17 (Fig. 1). Every adult ends up with a unique microbiota configuration, even identical twins that are raised in the same household18. Therefore, although microbiota assembly is not yet fully understood, adult microbial communities seem to be predominantly shaped by prior stochastic environmental exposures, as well as multiple other factors such as diet, antibiotic therapy and host genetics, with direct ‘inheritance’ from the mother at birth playing a similarly lesser role.
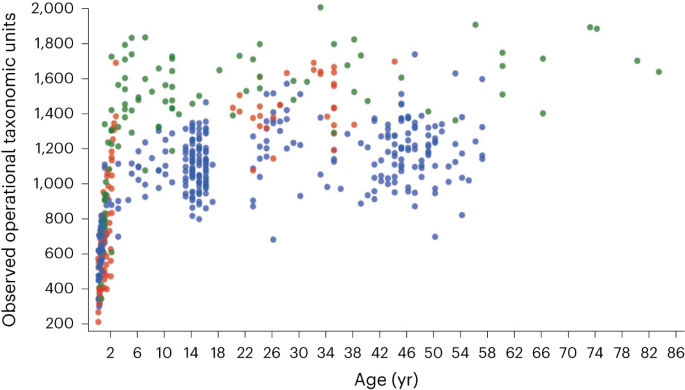
Diversity (as assessed using number of observed operational taxonomic units) dramatically increases during the first few years of life, particularly after weaning, before beginning to plateau in childhood. This pattern is observed across individuals living in different geographical locations: Malawi (red), Venezuela (green) and the United States (blue). Figure adapted with permission from ref. 17, Springer Nature Ltd.
“Most diseases are characterized by a pathobiome”
It has become increasingly common to read claims in the literature that most diseases are caused by a ‘pathobiome’. This is loosely defined as deleterious interactions between microbial communities and their host that lead to disease. This term is unfortunately overly simplistic and inherently flawed. Microorganisms and their metabolites are neither ‘good’ nor ‘bad’, they merely exist. Their impacts on us as hosts are heavily dependent on context. Microorganisms or metabolites that are deleterious in one context may cause no harm in another. As examples, Clostridioides difficile can be carried asymptomatically throughout life, and only cause problems in older age when the host is immunocompromised and treated with antibiotics19. Similarly, a strain of E.coli may be relatively harmless in the colon, but cause a urinary tract infection if it invades the urethra20. As a result, the term pathobiome remains vague and lacking in the precision required to be truly useful in clinical practice.
It is true, however, that numerous human conditions have been shown to correlate with alterations in microbiota composition. This is sometimes referred to as ‘dysbiosis’, which is also a vague term with limited clinical applicability21. It is very likely that this may contribute to disease progression in some conditions, including inflammatory bowel diseases22,23, however, such alterations are rarely consistent and the microbiota is hugely variable between individuals, both in health and disease. This makes it extremely difficult to identify gut microbiota configurations with the required specificity and reproducibility for clinical practice24. In addition, correlating gut microbiome changes with systemic markers or data is fraught with challenges. This often fails to account for confounders such as age, body mass index (BMI), sex and medication, factors such as microbial community interactions or for changes that occur as a result of immunological, metabolic or other functional changes in the host rather than being directly causal (Fig. 2). Attempts to define ‘tipping points’ at which changes in microbiome composition definitively influence disease progression have so far largely failed to generate a clear consensus due to a lack of consistency between different studies. It is, therefore, a leap that is not yet evidence based to conclude that a characteristic pathobiome has a role in ‘most’ diseases.
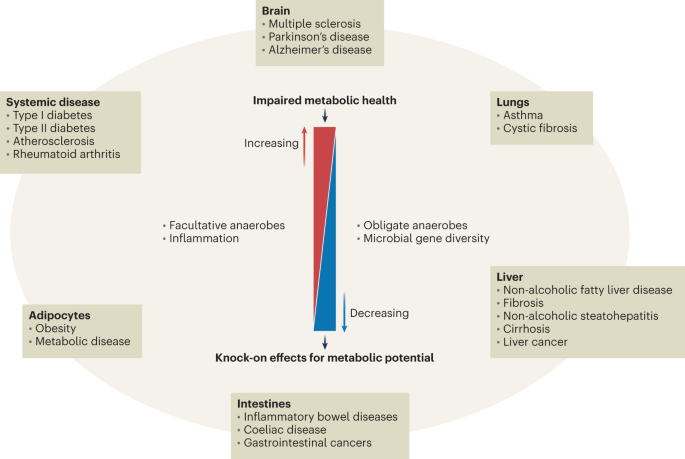
Changes in faecal microbiota have been associated with a range of diseases in humans. Interestingly, despite the diverse nature of these conditions, and the organs they affect, there are some broadly common recurring microbiota features, such as reduced diversity and increases in facultative anaerobes like Enterobacteriaceae. One common theme amongst these conditions is that they often result in increased levels of inflammation, at local and systemic levels. Such inflammation can, in turn, deplete the gut microbiota (and consequently microbial gene diversity), and allow facultative anaerobes such as Enterobacteriaceae to proliferate. This directly impacts the metabolic output of the microbiota, and its interactions with the host. Additionally, there are other host factors that contribute to disease and gut microbiota composition, such as age, BMI and medication, as well as host metabolism and immune response. This makes it very difficult to distinguish cause from effect in correlation-based studies.
“The Firmicutes:Bacteroidetes ratio is altered in obesity”
Related to the previous section, this commonly used but erroneous claim stems primarily from rodent-based research, and from findings in single, or under-powered, human studies. However, as with many other studies that report links between specific microbiota profiles and disease, reproducibility is poor. Indeed, there have now been at least three meta-analyses reporting that this finding is inconsistent between human studies, and that there are, in fact, no reproducible microbial taxonomic signatures of obesity in humans25,26,27.
This misconception also reflects an unhelpful tendency to examine sequence-based microbiota profiles at very broad taxonomic levels, such as phyla. Although this is appealing from a data simplification point of view, it fails to incorporate the huge and inherent variability within individual phyla. To draw a crude analogy, humans, birds, fish, reptiles and even sea squirts are all members of the phylum Chordata, yet clearly have very different physiologies, lifestyles and impacts on their environments.
Moreover, this claim was also based on relative-abundance-based DNA sequence data. Compositional data are still useful, and can correlate well with absolute quantifications obtained with techniques such as qPCR28,29. However, some studies have suggested that relative-abundance-based correlations can lose significance when absolute microbial abundances are also factored into analyses9. Moving forwards, increased incorporation of absolute quantification data may help to make conclusions based on compositional analyses more robust.
“The gut microbiome is functionally redundant”
This claim derives from studies showing that, whereas the taxonomic composition of human metagenomes can vary hugely, functional gene prediction profiles remain remarkably consistent. We contend that this is at least partly artefactual, as these functional comparisons are typically carried out after discarding the large proportion of metagenomic data that does not map to reference databases30. Much of what does map to those databases is likely to be derived from common housekeeping and/or well-characterized genes, which are found across many different bacteria and are also relatively well represented in reference databases. These comparative analyses therefore fail to accurately capture specialist, or less well-characterized, functions. As such, the truth is more nuanced. Although there are important functionalities that are conserved across many different human microbiota species, such as short-chain fatty acid production29, there are many key functions that are only carried out by a relatively small number of microbiota species. Examples include oxalate31 and resistant-starch32 degradation. In the absence of key species, functionalities such as these may not necessarily be fully replaced by other members of the microbiota.
“Sequencing is unbiased”
Although sequence-based methods have been transformative for microbiome research, they are not perfect. Biases can be introduced at every step of sequence-based studies, from sample collection and storage, through laboratory-based steps such as DNA extraction, to choice of bioinformatic pipelines and reference databases used to analyse the data33. Comparisons of sequence-based versus culture-based studies of the microbiota have shown that sequence-based approaches completely failed to detect some species that were only recovered using traditional culturing methods34. Modern sequence-based approaches are hugely powerful but, like all techniques, they are not unbiased. For optimal interpretation of results, it is important to be aware of the inherent limitations of any given method.
“We need standardized methodologies”
This opinion is prevalent in the microbiome field and is sensibly grounded in a desire to make it easier and more robust to compare results from different studies. However, as outlined above, there are no methodologies that are perfect, and all are biased in some way. If everyone in the world is using the same method, then everyone is equally blind to the limitations of that particular approach. There is also the problem of deciding which protocol everyone should use. For example, comparisons of results from the Human Microbiome Project with the MetaHIT project showed stark differences in microbiome profiles and indicated that the Human Microbiome Project protocol was less effective at extracting DNA from eukaryotes and Gram-positive bacterial lineages35. The truth is that the ‘best’ method fundamentally depends on the underlying structure of the microbial community in a given sample and this can vary hugely between individuals and between body sites. For these reasons we argue, as others have, that optimization and verification of sequence-based results with additional approaches are preferable to asking everyone to adopt the same method36. An additional advantage of multi-faceted studies using different methods and research platforms is that they can enable more mechanistic understandings of associations between microorganisms and host phenotypes37,38,39. Increased transparency when reporting methodology choices would be helpful for comparing results from different studies. The recently published STORMS (Strengthening the Organization and Reporting of Microbiome Studies) guidelines40, for example, could greatly aid this process if adopted widely.
“Most of the human microbiota is ‘unculturable’”
The adoption of high-throughput sequence-based technologies has also been mirrored by claims from some quarters that these methods must be used because most human-associated microorganisms cannot be cultivated in the laboratory. In fact, a reasonably large proportion of the bacterial and archaeal component of our microbiota has already been cultured41 (viruses and fungi remain under-represented), with pioneering work from as early as the 1970s establishing the cultivability of a broad diversity of species from the human gut microbiota42. Many more species continue to be cultured as laboratory-based efforts have increased throughput43,44. This implies that current gaps in culture collections are at least in part attributable to a lack of previous effort rather than an inherent ‘unculturability’. Although cultivation is undeniably labour intensive, has its own biases and often requires expensive specialist equipment and media, there are clear advantages to having microorganisms in culture. These include enabling mechanistic experiments, verifying genomic predictions, and developing them as novel therapeutics45. Given the importance of continued cultivation-based work for the progression of microbiome research, it is gratifying that this myth has become less prevalent in recent years following the publication of the aforementioned high-impact studies that demonstrate it to be false. However, it serves as an excellent example of how previously widely accepted dogma is sometimes simply not true. There are important lessons for many other myths and misconceptions that have yet to become as widely rejected.
Conclusions
The microbiome field is broad, and there are many other controversial topics that might also have been included here. However, knowledge is still evolving on many of these; consequently, we have largely focused on concepts where we believe there is a strong evidence base for rejecting myths and misconceptions. Although some of the points above may seem trivial, we argue that the accuracy of details such as these matters. If we are consistently repeating falsehoods about minor details, can our accuracy be relied upon when covering more important matters? We hope that, by illustrating just a few examples of microbiome myths and misconceptions, we can draw increased attention to the potential problems of over-simplification and insufficient critical assessment in the microbiome literature.
Given the many potential health impacts, the huge amount of funding and the keen public interest in microbiomes, rejection of unfounded assertions is crucial if we wish to avoid expending finite resources researching unproductive avenues and undermining public confidence in our conclusions.